|
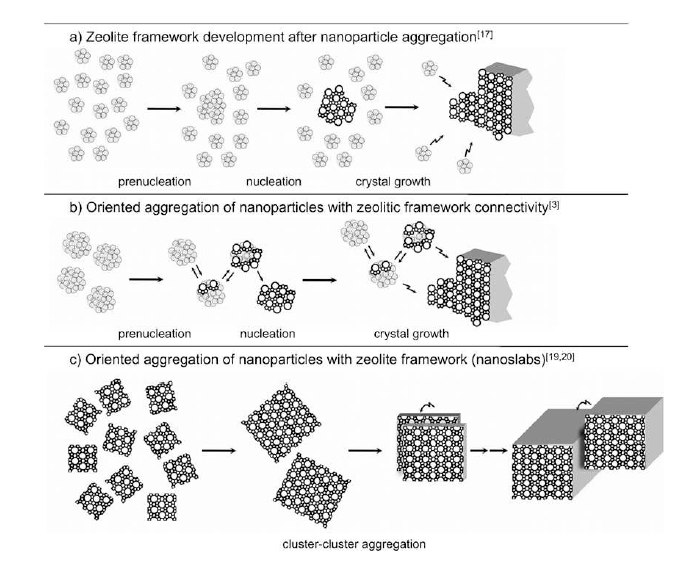
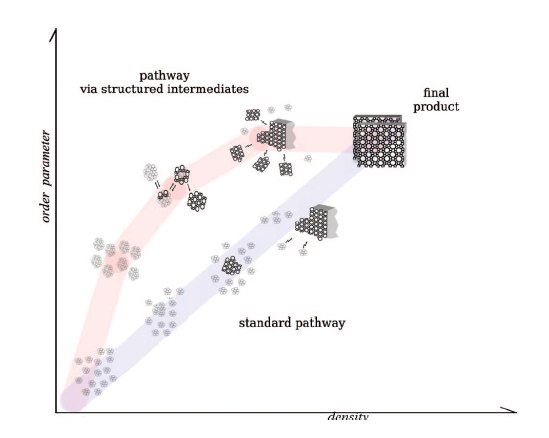
In concentrated clear sol prepared from tetraethylorthosilicate, tetrapropylammonium hydroxide, and water suitable for crystallization of Silicalite-1 zeolite, the main part of the silica is present in nanoparticles. The nature of these nanoparticles and their evolution during the induction period and the stage of early crystal growth was investigated via dissolution experiments in the presence of excess TPAOH. The dissolution process was monitored in situ using static and dynamic light scattering (SLS/DLS) and synchrotron small-angle X-ray scattering (SAXS). The complete dissolution of an individual nanoparticle was observed to occur in one step. Dissolution transformed a nanoparticle into a cluster of silicate oligomers. Larger grown nanoparticles dissolved slower. Exponential dissolution rate constants scaled inversely proportional with the volume of the nanoparticle's silica core. This experimentally observed dissolution behavior was modeled by assuming that a nanoparticle dissolved to oligomers via a series of partially dissolved nanoparticles that correspond to metastable intermediate states of increasing free energy. The resulting free energy barrier that has to be overcome by a dissolving nanoparticle could be derived from the experimental input. The idealized free energy profile provided a qualitative explanation for the apparent instantaneous disintegration of entire nanoparticles. |
|
|
Heat and mass transfer in nanodevices depends much on the geometry due to the strong influence of curvature on interfacial properties, such as the Kapitza resistance. We present a method which combines nonequilibrium square gradient theory and nonequilibrium molecular dynamics simulations to obtain the coefficients in a curvature expansion of the interface transfer coefficients. The expansion can be used directly to describe heat and mass transfer in complex nano-geometries. As examples of complex nano-geometries we consider an oblate spheroidal droplet, a prolate spheroidal bubble, and a toroidal bubble. Depending on the sign and magnitude of the curvature, transfer is enhanced or reduced significantly. The presented method is applicable to many types of interfaces and substances, and we expect it to contribute to the understanding and design of future nanodevices. |
|
We introduce a method to evaluate simultaneously the reaction rate constant and the free energy profile of a process in a complex environment. The method employs the partial path transition interface sampling technique we recently developed for the calculation of rate constants in diffusive systems. We illustrate the applicability of the technique by studying a simple dimer in a repulsive fluid, and show that the free energy can be obtained at no additional computational cost. |
|
|
We review two recently developed efficient methods for calculating rate constants of processes dominated by rare events in high-dimensional complex systems. The first is transition interface sampling (TIS), based on the measurement of effective fluxes through hypersurfaces in phase space. TIS improves efficiency with respect to standard transition path sampling (TPS) rate constant techniques, because it allows a variable path length and is less sensitive to recrossings. The second method is the partial path version of TIS. Developed for diffusive processes, it exploits the loss of long time correlation. We discuss the relation between the new techniques and the standard reactive flux methods in detail. Path sampling algorithms can suffer from ergodicity problems, and we introduce several new techniques to alleviate these problems, notably path swapping, stochastic configurational bias Monte Carlo shooting moves and order-parameter free path sampling. In addition, we give algorithms to calculate other interesting properties from path ensembles besides rate constants, such as activation energies and reaction mechanisms. |
|
|
We briefly review simulation schemes for the investigation of rare transitions and resume the recently introduced transition interface sampling, a method in which the computation of rate constants is recast into the computation of fluxes through interfaces dividing the reactant and product state. |
|
We derive a novel efficient scheme to measure the rate constant of transitions between stable states separated by high free energy barriers in a complex environment within the framework of transition path sampling. The method is based on directly and simultaneously measuring the fluxes through many phase space interfaces and increases the efficiency with at least a factor of 2 with respect to existing transition path sampling rate constant algorithms. The new algorithm is illustrated on the isomerization of a diatomic molecule immersed in a simple fluid. In this paper the Transition Interface Sampling (TIS) method was introduced, an important method that is nowadays widely used. Several other popular methods are based on TIS such as PPTIS, RETIS, and Forward Flux Sampling. |
|
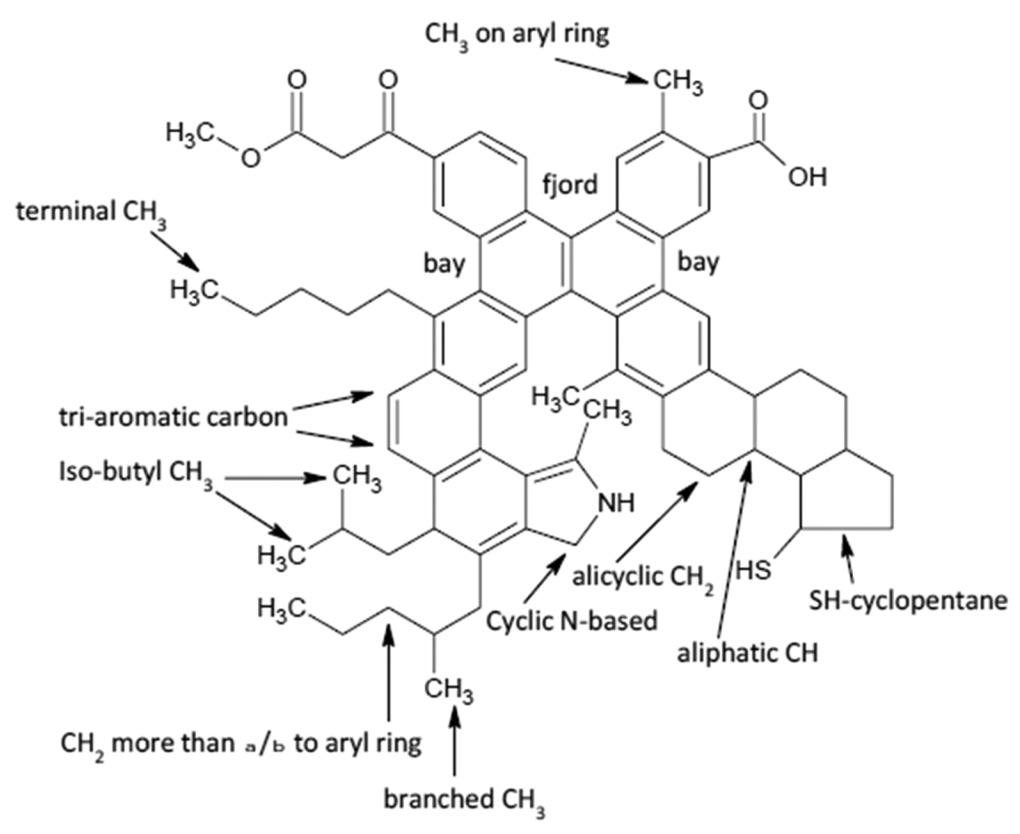
Due to their complex chemical structure, identification and characterization of asphaltenes remain a big challenge for researchers in the oil industry. In the current contribution, solution-state two-dimensional (2D) nuclear magnetic resonance (NMR) spectra of the asphaltenes, obtained from either heavy or light crude oil samples, provide information on the chemical structures of asphaltenes at the molecular level. Two-dimensional 1H–13C heteronuclear NMR measurements are particularly useful for differentiating among several structural environments whose signals overlap in the one-dimensional 13C spectra. Both, 2D 1H–13C heteronuclear and 1H–1H homonuclear NMR spectra indicate short- and long-range interactions between different functional groups of the asphaltene samples, revealing even “bay”- and “fjord”-type structures. Based on the NMR correlations between different protons and carbons with different chemical environments, we propose various chemical structures (polyaromatic cores with aliphatic chains, porphyrin derivatives, organic salts). Furthermore, through molecular dynamics simulations, we have obtained Hildebrand solubility parameters of different asphaltene solvents and of two experimental recovered asphaltene structures. Our calculated solubilities are consistent with previous experimental and simulation works. |
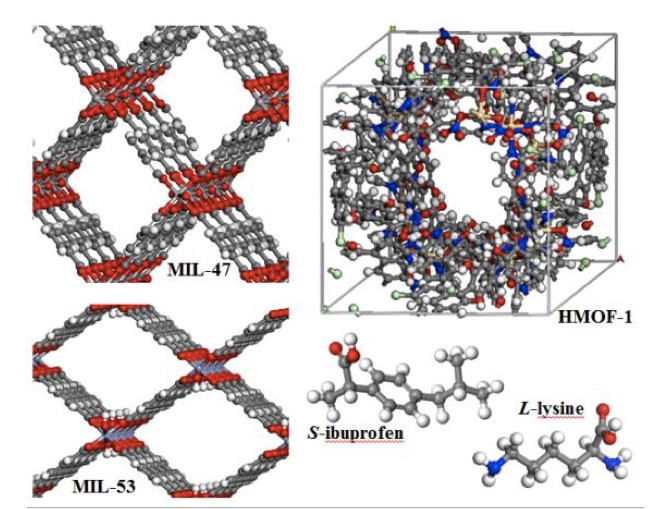
|
This study reveals efficient enantiomeric separation of bioactive molecules in liquid phase. Chiral structure H-MOF-1 separates racemic mixtures whereas heteroselectivity is observed for scalemic mixtures of ibuprofen using non-chiral MIL-47 and MIL-53. Lysine enantiomers are only separated by H-MOF-1. These separations are controlled by the tight confinement of the molecules. |

|
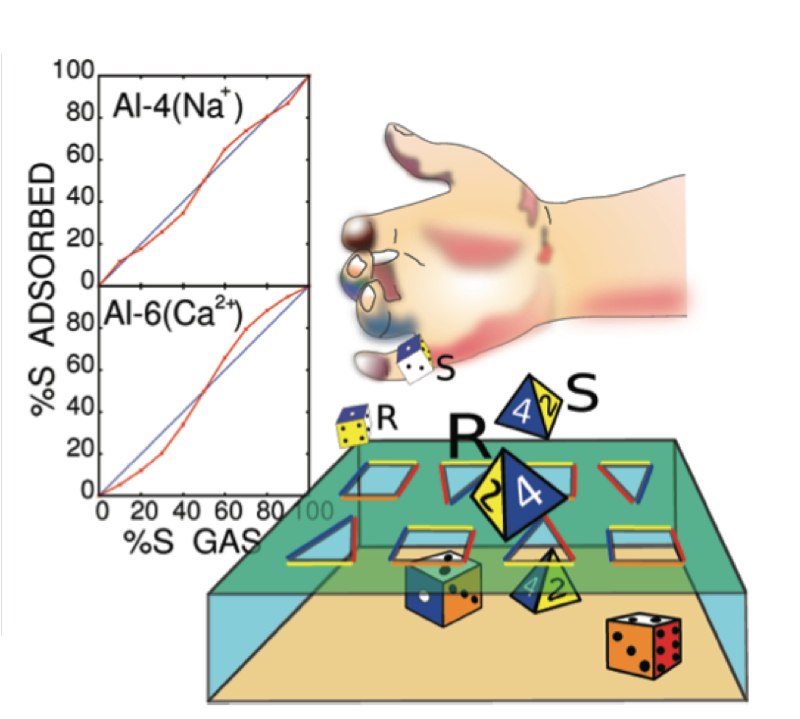
Using computer simulations, we have studied the adsorption of racemic and scalemic mixtures of 4-ethyl-4-methyloctane molecules in aluminum-substituted MFI zeolites containing positively charged ions. Our results show that aluminum distribution affects the shape of the enantiopure adsorption isotherms, which is contrary to previously established results on linear alkanes. In addition, we examined the enantiospecific adsorption at maximum loading conditions. For enantiopure and racemic mixtures, the fraction of S- and R-enantiomers in the adsorbed phase is identical to that of the gas phase. However, when the external gas contains a scalemic mixture, the dominant enantiomeric fraction might either increase or decrease upon adsorption, depending on the aluminum distribution and on the type of cations. We provide a theoretical explanation for both types of adsorption characteristics. Our findings open perspectives for new approaches of enantioseparation that do not require a chiral adsorbent. |
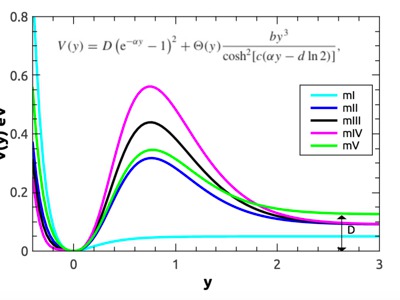
|
Although previously developed mesoscopic DNA models have successfully reproduced thermodynamic denaturation data, recent studies show that these overestimate the rate of denaturation by orders of magnitude. Using adapted Peyrard-Bishop-Dauxois (PBD) models, we calculated the denaturation rates of several DNA hairpins and made comparison with experimental data. We show that the addition of a barrier at the onsite potential of the PBD model gives a more accurate description of the unzipping dynamics of short DNA sequences. The new models provide a refined theoretical insight on the dynamical mechanisms of unzipping which can have implications for the understanding of transcription and replication processes. Still, this class of adapted PBD models seems to have a fundamental limitation which implies that it is not possible to get agreement with available experimental results on the dynamics of long DNA sequences and at the same time maintain the good agreement regarding its thermodynamics. The reason for this is that the denaturation rate of long DNA chains is not dramatically lowered by the additional barrier as the base-pairs that open are more likely to remain open, facilitating the opening of the full DNA molecule. Some care has to be taken since experimental techniques capable to study denaturation rates of long sequences seem not in agreement with other experimental data on short DNA sequences. Further research, both theoretical and experimental, is therefore needed to resolve these inconsistencies, which will be a starting point for new minimalistic models that are able to describe both thermodynamics as well as dynamics at a predictive level. |
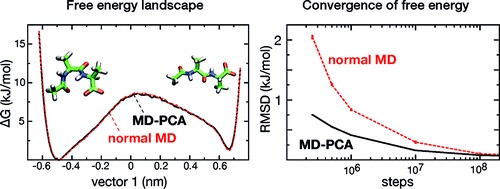
|
A molecular dynamics algorithm in principal component space is presented. It is demonstrated that sampling can be improved without changing the ensemble by assigning masses to the principal components proportional to the inverse square root of the eigenvalues. The setup of the simulation requires no prior knowledge of the system; a short initial MD simulation to extract the eigenvectors and eigenvalues suffices. Independent measures indicated a 6-7 times faster sampling compared to a regular molecular dynamics simulation. |
|
|
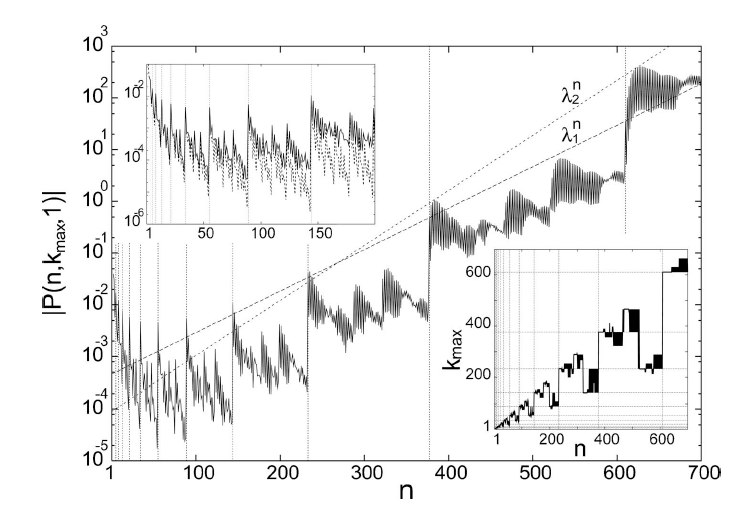
Incommensurate structures can be described by the Frenkel-Kontorova model. Aubry has shown that, at a critical value Kc of the coupling of the harmonic chain to an incommensurate periodic potential, the system displays the analyticity-breaking transition between a sliding and pinned state. The ground-state equations coincide with the standard map in non-linear dynamics, with smooth or chaotic orbits below and above Kc, respectively. For the standard map, Greene and MacKay have calculated the value Kc = 0.971635. Conversely, evaluations based on the analyticity breaking of the modulation function have been performed for high commensurate approximants. Here we show how the modulation function of the infinite system can be calculated without using approximants but by Taylor expansions of increasing order. This approach leads to a value Kc' = 0.97978, implying the existence of a golden invariant circle up to Kc' > Kc. This paradox was solved in later paper: Breakdown of Lindstedt expansion for chaotic maps, J. Math. Phys., 46, 102702, (2005), Guido Gentile, and Titus S. van Erp. |
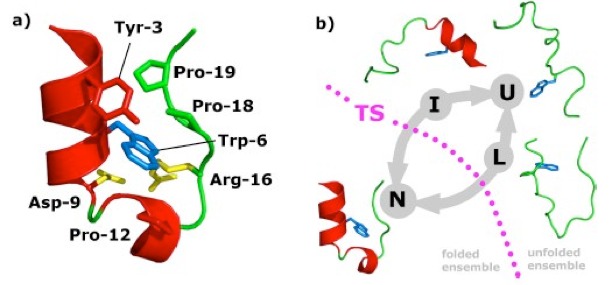
|
Numerically predicting rate constants of protein folding and other relevant biological events is still a significant challenge. We show that the combination of partial path transition interface sampling with the optimal interfaces and free-energy profiles provided by path collective variables makes the rate calculation for practical biological applications feasible and efficient. This methodology can reproduce the experimental rate constant of Trp-cage miniprotein folding with the same level of accuracy as transition path sampling at a fraction of the cost. |
|
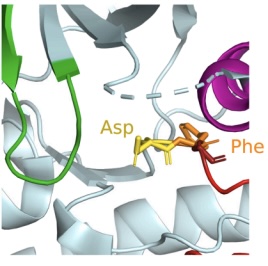
Predicting the kinetics of drug-protein interactions is crucial for understanding drug efficacy, particularly in personalized medicine, where protein mutations can significantly alter drug residence times. This study applies replica exchange transition interface sampling and its partial path variant to investigate the dissociation kinetics of imatinib from Abelson nonreceptor tyrosine kinase (ABL) and mutants relevant to chronic myeloid leukemia therapy. These path sampling methods offer a bias-free alternative to conventional approaches requiring qualitative predefined reaction coordinates. Nevertheless, the complex free energy landscape of ABL-imatinib dissociation presents significant challenges. Multiple metastable states and orthogonal barriers lead to parallel unbinding pathways, complicating convergence in transition interface sampling-based methods. Despite employing computational efficiency strategies such as asynchronous replica exchange, full convergence remained elusive. This work provides a critical assessment of path sampling in high-dimensional biological systems, discussing the need for enhanced initialization strategies, advanced Monte Carlo path generation moves, and machine learning-derived reaction coordinates to improve kinetic predictions of drug dissociation with minimal prior knowledge. |
|
|
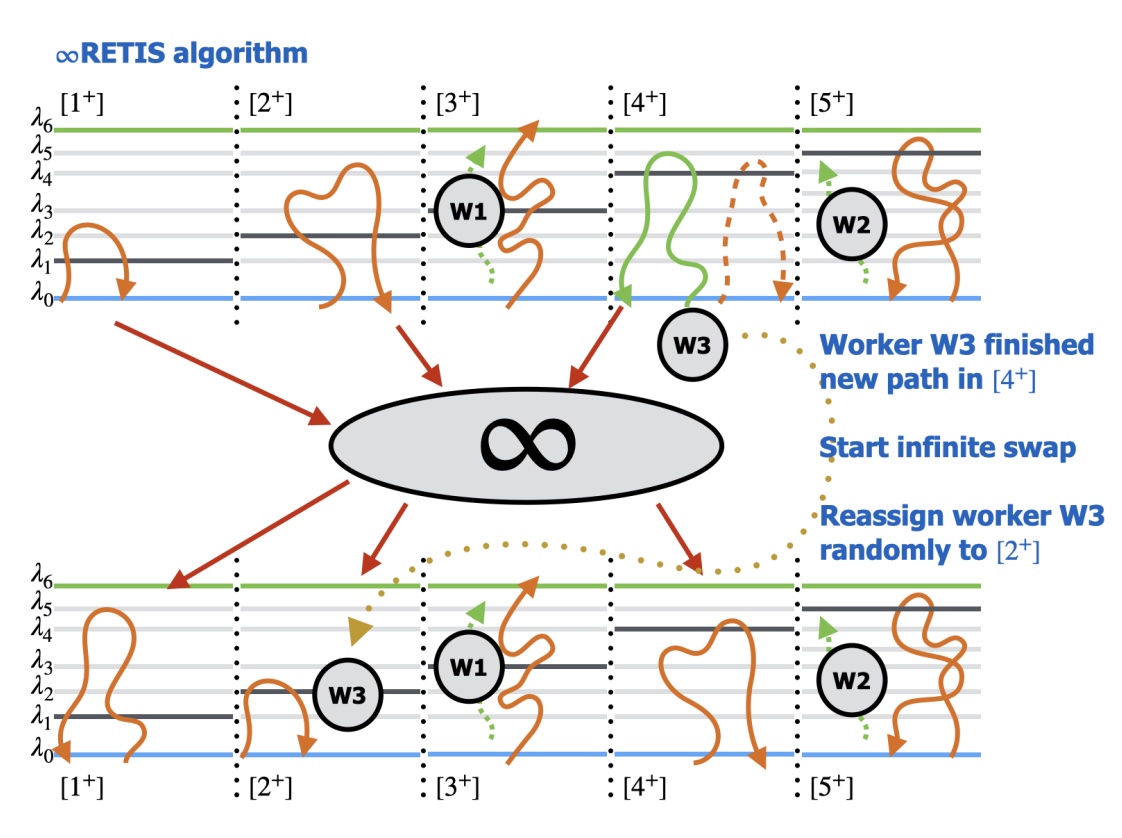
We implement ∞RETIS, the most advanced variant of the replica exchange transition interface sampling (RETIS) algorithm, which employs asynchronous swapping moves in the infinite swapping limit. This design enables highly efficient parallelization across CPUs and GPUs, resulting in a substantial acceleration in convergence. Using molecular dynamics (MD) simulations, we apply this method to investigate the membrane permeation of 5-aminolevulinic acid (5-ALA), a hydrophilic drug widely used in photodynamic therapy (PDT) and fluorescence-guided surgery. Key kinetic properties, including the permeability and mean first passage time, are computed from the resulting unbiased trajectories. Furthermore, the mechanistic details of 5-ALA permeation are explored, showing the impact of the 5-ALA orientation and its hydration by water molecules inside the membrane on whether a trajectory contributes to successful membrane crossing. By resolving the full kinetic mechanism of 5-ALA permeation through a phospholipid bilayer, this study showcases the power of ∞RETIS in addressing rare event dynamics in biologically and pharmaceutically relevant systems. |
|
Assessing kinetics in biological processes with molecular dynamics simulations remains a computational and conceptual challenge, given the large time and length scales involved. For kinetic transport of biochemical compounds or drug molecules, the permeability through the phospholipid membranes is a key kinetic property, but long timescales are hindering the accurate computation. Technological advances in high-performance computing therefore need to be accompanied by theoretical and methodological developments. In this contribution, the replica exchange transition interface sampling (RETIS) methodology is shown to give perspective toward observing longer permeation pathways. It is first reviewed how RETIS, a path-sampling methodology that gives in principle exact kinetics, can be used to compute membrane permeability. Next, recent and current developments in three RETIS aspects are discussed: several new Monte Carlo moves in the path-sampling algorithm, memory reduction by reducing pathlengths, and exploitation of parallel computing with CPU-imbalanced replicas. Finally, the memory reduction presenting a new replica exchange implementation, coined REPPTIS, is showcased with a permeant needing to pass a membrane with two permeation channels, either representing an entropic or energetic barrier. The REPPTIS results showed clearly that inclusion of some memory and enhancing ergodic sampling via replica exchange moves are both necessary to obtain correct permeability estimates. In an additional example, ibuprofen permeation through a dipalmitoylphosphatidylcholine membrane was modeled. REPPTIS succeeded in estimating the permeability of this amphiphilic drug molecule with metastable states along the permeation pathway. In conclusion, the presented methodological advances allow for deeper insight into membrane biophysics even if the pathways are slow, as RETIS and REPPTIS push the permeability calculations to longer timescales |
|
|
Several simulations strategies have emerged to predict the permeability of solutes across membranes, which is important for many biological or industrial processes such as drug design. The widespread inhomogeneous solubility-diffusion (ISD) model is based on the Smoluchowski equation and describes permeation as purely diffusive. The counting method, which counts membrane transitions in a long molecular dynamics (MD) trajectory, is free of this diffusive assumption, but it lacks sufficient statistics when the permeation involves high free energy barriers. Metadynamics and variations thereof can overcome such barriers, but they generally lack the kinetics information. The milestoning framework has been used to describe permeation as a rare event, but it still relies on the Markovian assumption between the milestones. Replica Exchange Transition Interface Sampling (RETIS) has been shown to be an effective method for sampling rare events while simultaneously describing the kinetics without assumptions. This paper is the first permeation application of RETIS on an all-atom lipid bilayer consisting of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) to compute the entrance, escape and complete transition of molecular oxygen. Conventional MD was performed as a benchmark, and the MD rates from counting were converted to rate constants, giving good agreement with the RETIS values. Moreover, a correction factor was derived to convert the collective order parameter in RETIS, which was aimed to improve efficiency, to a single-particle order parameter. With this work, we showed how the exact kinetics of drug molecules permeation can be assessed with RETIS even if the permeation is truly a rare event or if the permeation is non-Markovian. RETIS will therefore be a valuable tool for future permeation studies. |
|
Despite its approximative nature, the Langmuir theory has shown to be a very successful approach to describe experimental adsorption isotherms. Langmuir kinetics is based on systems of non- interacting particles that can transfer from the gas phase to the adsorbed phase with a transition flux that depends both on the gas pressure and surface coverage. Recent molecular simulation results suggest, however, that some systems can have isotherms that are apparently Langmuirian while the kinetics are not. This remarkably result seems to question the interpretation of innumerous adsorption experiments. The observed anomalous kinetics were described by thermodynamic rate equations giving exactly the same isotherms. Unidirectional rates, as obtained from mesoscopic non- equilibrium theory, correct for the non-ideality of matter using activities instead of concentrations and seem to suggest that fluxes from phase A to another phase B only depends on the properties of phase A alone. In this article we show, however, that the theories and simulations are actually consistent when the following two points are taken into account. The first point is methodological and related to how one should count crossing events considering the presence of possible correlations. The second point is theoretical and related to the microscopic link between the Langmuir and thermodynamic rate theory. Specifically, we show how to define diffusion and activity coefficients at the border of the gas/solid interface. If both points are taken into account, there is neither a contradiction between both theories, nor with the molecular simulation results. This article was published in a new open-access journal and served as inauguration article of Prof. Signe Kjelstrup as Associate Editor in the section Physical Chemistry and Chemical Physics. |
|
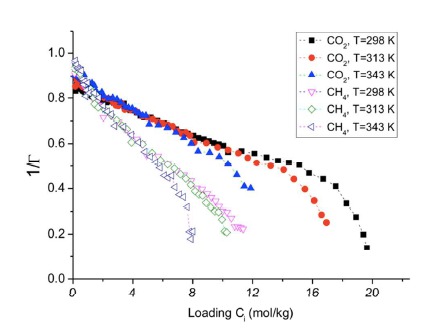
Thermodynamic equilibrium for adsorption means that the chemical potential of gas and adsorbed phase are equal. A precise knowledge of the chemical potential is, however, often lacking, because the activity coefficient of the adsorbate is not known. Adsorption isotherms are therefore commonly fitted to ideal models such as the Langmuir, Sips or Henry models. We propose here a new procedure to find the activity coefficient and the equilibrium constant for adsorption which uses the thermodynamic factor. Instead of fitting the data to a model, we calculate the thermodynamic factor and use this to find first the activity coefficient. We show, using published molecular simulation data, how this procedure gives the thermodynamic equilibrium constant and enthalpies of adsorption for CO2(g) on graphite. We also use published experimental data to find similar thermodynamic properties of CO2(g) and of CH4(g) adsorbed on activated carbon. The procedure gives a higher accuracy in the determination of enthalpies of adsorption than ideal models do. |
|
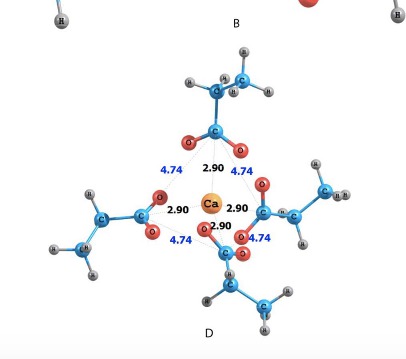
This work, interactions between carboxylate ions and calcium or sodium ions are investigated via Density Functional Theory (DFT). Despite the ubiquitous presence of these interactions in natural and industrial chemical processes, few DFT studies on these systems exist in the literature. Special focus has been placed on determining the influence of the multibody interactions (with up to 4 carboxylates and one metal ion) on an effective pair-interaction potential, such as those used in molecular mechanics (MM). Specifically, DFT calculations are employed to quantify an effective pair-potential that implicitly includes multibody interactions in order to construct potential energy curves for carboxylate-metal ion pairs. The DFT calculated potential curves are compared to a widely used molecular mechanics force field (OPLS-AA). The calculations indicate that multibody effects do influence the energetic behavior of these ionic pairs and the extent of this influence is determined by a balance between (a) charge transfer from the carboxylate to the metal ions which stabilizes the complex, and (b) repulsion between carboxylates which destabilizes the complex. Additionally, the potential curves of the complexes with 1 and 2 carboxylates and one counter ion have been examined to higher separation distance (20 Å) by the use of relaxed scan optimization and constrained density functional theory (CDFT). The results from the relaxed scan optimization indicate that near the equilibrium distance, the charge transfer between the metal ion and the deprotonated carboxylic acid group is significant and leads to non-negligible differences between the DFT and MM potential curves, especially for calcium. However, at longer separati on distances the MM calculated interaction potential functions converge to those calculated with CDFT, effectively indicating the approximate domain of the separation distance coordinate where charge transfer between the ions is occurring. |
|
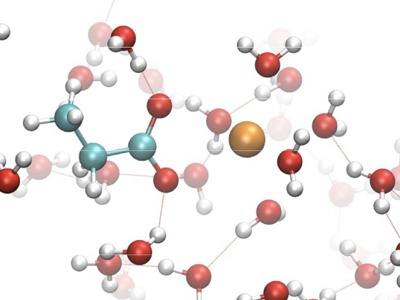
The interaction between a carboxylate anion (deprotonated propanoic acid) and the divalent - Mg2+, Ca2+, Sr2+, Ba2+ metal ions is studied via ab initio molecular dynamics. The main focus of the study is the selectivity of the carboxylate - metal ion interaction in aqueous solution. The interaction is modelled by explicitly accounting for the solvent molecules on a DFT level. The hydration energies of the metal ions along with their diffusion and mobility coefficients are determined and a trend correlated with their ionic radius is found. Subsequently, a series of 16 constrained molecular dynamics simulations for every ion are performed and the interaction free energy is obtained from thermodynamic integration of the forces between the metal ion and the carboxylate ion. The results indicate that the magnesium ion interacts most strongly with the carboxylate, followed by calcium, strontium and barium. Since the interaction free energy is not enough to explain the selectivity of the reaction observed experimentally, more detailed analysis is performed on the simulation trajectories in order to understand the steric changes in the reaction complex during dissociation. The solvent dynamics appear to play an important role during the dissociation of the complex and also in the observed selectivity behaviour of the divalent ions. |
|
|
We have studied the ground state and phonons of a generalized Frenkel Kontorova model with a quasiperiodic substrate potential. We have recently shown that, depending on the three mutually incommensurate, relevant lengths, a sliding mode below a critical coupling can either occur or be absent although a structural transition takes place in both cases. Besides, we observe a transition to localized phonon states. Here we investigate the structural and localization transitions in terms of a nonlinear discrete map. |
|
We show that the static and dynamic properties of the Frenkel-Kontorova (FK) model drastically change when an incommensurate harmonic is added to the periodic potential. Our model consists of a harmonic chain with spacing l on a quasiperiodic substrate potential of the form, where the three relevant lengths l, 1, and tau-1 are chosen to be mutually incommensurate. Within this model we identify two classes of behavior. One presents a sliding mode up to an analyticity breaking, as in the FK model, and another is pinned for any strength of the additional harmonic. Besides, we show that in all cases if 0 or 1, localization of phonons exists beyond a critical value of the potential strength. |
|
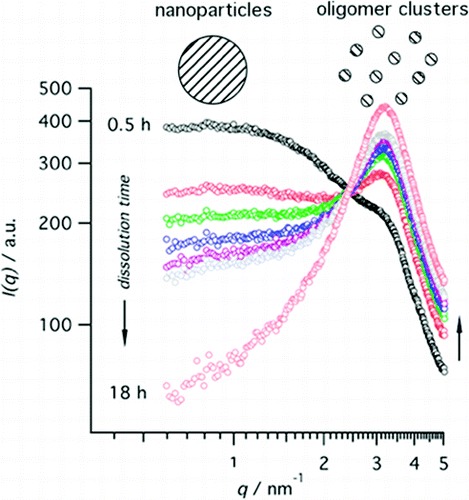
Colloidal silicalite-1 zeolite was crystallized from a concentrated clear sol prepared from tetraethylorthosilicate (TEOS) and aqueous tetrapropylammonium hydroxide (TPAOH) solution at 95C. The silicate speciation was monitored by using dynamic light scattering (DLS), synchrotron small-angle X-ray scattering (SAXS), and quantitative liquid-state 29Si NMR spectroscopy. The silicon atoms were present in dissolved oligomers, two discrete nanoparticle populations approximately 2 and 6 nm in size, and crystals. On the basis of new insight into the evolution of the different nanoparticle populations and of the silicate connectivity in the nanoparticles, a refined crystallization mechanism was derived. Upon combining the reagents, different types of nanoparticles (ca. 2 nm) are formed. A fraction of these nanoparticles with the least condensed silicate structure does not participate in the crystallization process. After completion of the crystallization, they represent the residual silicon atoms. Nanoparticles with a more condensed silicate network grow until approximately 6 nm and evolve into building blocks for nucleation and growth of the silicalite-1 crystals. The silicate network connectivity of nanoparticles suitable for nucleation and growth increasingly resembles that of the final zeolite. This new insight into the two classes of nanoparticles will be useful to tune the syntheses of silicalite-1 for maximum yield. |
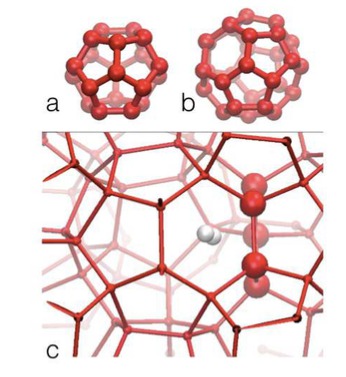
|
The transport of gas molecules in hydrates is presently poorly understood. In sII structured hydrates with hydrogen guests there is, for instance, a mismatch between experimental and computed values for diffusion constants. We provide an explanation for the experimentally observed diffusion rates, using DFT-based molecular dynamics simulations at 100 K. By considering the effect of cage occupancy, as well as the flexibility of the water lattice, we show that barriers for hydrogen hopping between cages, can approach values as low as 5 kJ/mol, which is very close to experimental values. |
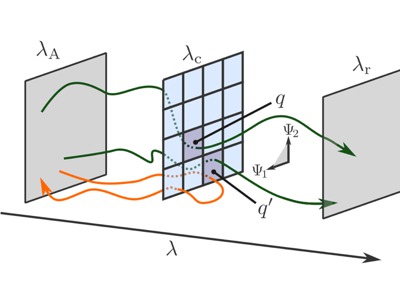
|
We introduce an approach to analyze collective variables regarding their predictive power for a reaction. The method is based on already available path sampling data produced by for instance transition interface sampling or forward flux sampling which are path sampling methods used for efficient computation of reaction rates. By a search in collective variable space a measure of predic- tiveness can be optimized and, in addition, the number of collective variables can be reduced using projection operations which keep this measure invariant. The approach allows testing hypotheses on the reaction mechanism, but could in principle also be used to construct the phase space com- mittor surfaces without the need of additional trajectory sampling. The procedure is illustrated for a one-dimensional double well potential, a theoretical model for an ion-transfer reaction in which the solvent structure can lower the barrier, and an Ab Initio molecular dynamics study of water auto-ionization. The analysis technique enhances the quantitative interpretation of path sampling data which can provide clues on how chemical reactions can be steered in desired directions. |
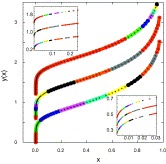
|
Brunauer-Emmett-Teller (BET) theory is a popular method to interpret nitrogen adsorption isotherms for determining the specific surface area. Besides the monolayer capacity nmnm, the BET equation depends on one single additional parameter C that is adjusted to fit the experimental adsorption curves. BET analysis is key in surface characterization and is widely applied even when the underlying assumptions are not strictly justified. For example, disregarding its weaker physical interpretation, the BET surface area is generally accepted as an important characterization of microporous materials. However, whereas the accuracy of the adsorption techniques has tremendously increased in recent years, experimentally derived BET values do not yet meet the same standards. The reason for this is due to some intrinsic ambiguities in its fitting procedure. In this letter, we propose a procedure that is uniquely defined, given a certain pressure range, and is independent of arbitrary experimental details such as the distribution of data points. It puts an equal weight on both monolayer and multilayer formation and gives an additional reproducible measure that is related to the inherent deviation from the BET behavior. |
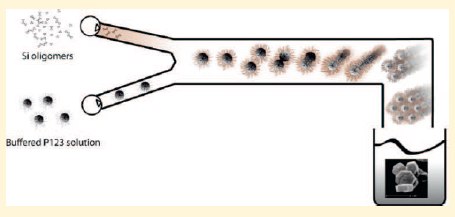
|
Hexagonally ordered mesoporous silica coined COK-12 was synthesized in a continuous process by combining streams of sodium silicate and citric acid/sodium citrate buffered solution of (ethylene oxide)20-propylene oxide)70-ethylene oxide)20 triblock copolymer (Pluronic P123) from separate reservoirs. COK-12 precipitated spontaneously upon combining both streams at nearly neutral pH and ambient temperature. Stable intermediates of the COK-12 formation process could be prepared by limiting sodium silicate addition. Investigation of these intermediates using small-angle X-ray scattering revealed COK-12 formed via an assembly process departing from spherical uncharged core-shell P123-silica micelles. The sterical stabilization of these micelles decreased upon accumulation of silicate oligomers in their shell. Aggregation of the spherical micelles led to cylindrical micelles, which aligned and adopted the final hexagonal organization. This unprecedentedly fast formation of P6m ordered mesoporous silica was caused by two factors in the synthesis medium: the neutral pH favoring uncharged silicate oligomers and the high salt concentration promoting hydrophobic interactions with surfactant micelles leading to silica accumulation in the PEO shell. The easy continuous synthesis process is convenient for large-scale production. The platelet particle morphology with short and identical internal channels will be advantageous for many applications such as pore replication, nanotube or fiber growth, catalytic functionalization, drug delivery, film and sensor development, and in nano dyes as well as for investigation of pore diffusion phenomena. |
|
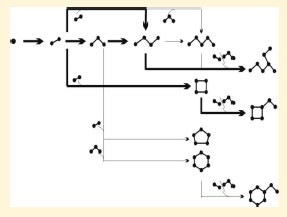
The initial molecular steps of the acid-catalyzed silica sol-gel process de-parting from tetraethylorthosilicate (TEOS) were investigated by in situ 29Si NMR and UV-Raman spectroscopy. The use of a substoichiometric H2O:TEOS molar ratio (r-value 0.2-1.2) slowed the silicate oligomerization reaction and allowed unraveling the initial steps of silica condensation. Molecular modeling confirmed Raman signal and 29Si NMR shift assignment. A comprehensive listing of all Raman and 29Si NMR assignments is provided, including unique Raman assignments of cyclosilicates and the linear tetramer. The combination of experiment and modeling allowed an analysis of the reaction kinetics. The derived kinetic model and the experimental observation both revealed that the H2O:TEOS molar ratio had a strong influence on the reaction kinetics but not on the reaction pathways. The multianalytical approach led to development of an oligomerization scheme. As dominant oligomerizations, chain growth, cyclodimerization, and branching were identified. Under the investigated conditions, chains did not grow longer than pentamer, and ring sizes were limited to 6-rings. Chains of 4 Si atoms and 4-rings were abundant species. Branched rings and chains were formed by attachment of dimers and trimers. Gelation proceeded from branched 4-rings and branched chains with limited hydroxyl functionalities. |
|
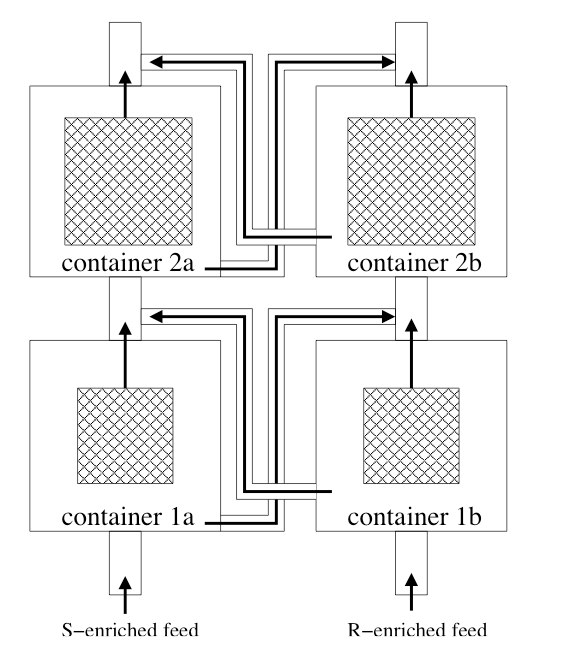
Chiral amplification is a known phenomenon in crystallization. Recent atomistic simulations have shown that amplification can also occur in equilibrium processes when a mixtures of chiral molecules adsorb in metal-substituted zeolites that are not chiral by themselves. In this article we investigate how this process could be exploited for enantiopurification and separation in a pressure-swing adsorption process. For this purpose, we develop a mathematical model that reproduces the main features of the full-atom simulations and allows us to study the impact of several parameters on the enantioselectivity on a macroscopic scale. Besides numerical solutions of this model, we provide some analytic results which suggests that the enantioselective materials prohibit a phase transition as a function of temperature. Below a certain critical temperature, the enantiomeric content of the adsorbed phase becomes a discontinuous function of the enantiomeric gas content. This has the following consequences: (i) the adsorbed phase in racemic conditions becomes unstable, (ii) a sudden increase in efficiency of the purification process, and (iii) the possibility to not only purify but also fully separate chiral mixtures using a parallel batch process in which both lines use an input that is enriched with opposite enantiomer. |
|
We devise an efficient Monte Carlo scheme to study the adsorption of a multicomponent gas in a nanoporous material. The configurational bias move is extended by a novel replica exchange procedure where the configurations of the different simulations describing one particular gas content are being swapped. For chiral mixtures, the efficiency can be further improved using the chiral inversion move. The method is demonstrated for an Ising-type model and a complicated realistic zeolite system. |
|
Minority excluded: If mixtures of chiral compounds are adsorbed in Al-substituted MFI zeolite containing nonframework cations, a process occurs that ultimately might be used for enantioseparation. If one enantiomer is present in excess it will organize the nearby cations, which in turn influence all other cations through long-range interaction, such that all adsorption sites become effectively chiral. This suppresses the minority enantiomer. |
|
|
The transition interface sampling (TIS) technique allows large free energy barriers to be overcome within reasonable simulation time, which is impossible for straightforward molecular dynamics. Still, the method does not impose an artificial driving force, but it surmounts the timescale problem by an importance sampling of true dynamical pathways. Recently, it was shown that the efficiency of TIS when calculating reaction rates is less sensitive to the choice of reaction coordinate than those of the standard free energy based techniques. This could be an important advantage in complex systems for which a good reaction coordinate is usually very difficult to find. We explain the principles of this method and discuss some of the promising applications related to zeolite formation. |
|
Solvation structures play an important role in aqueous chemistry involving protons. Subtle changes in the coordination of water molecules to a reacting species can be crucial for initiating a reactive event as is shown by van Erp and Meijer in a molecular simulation study of the proton-assisted hydration of ethylene. |
|
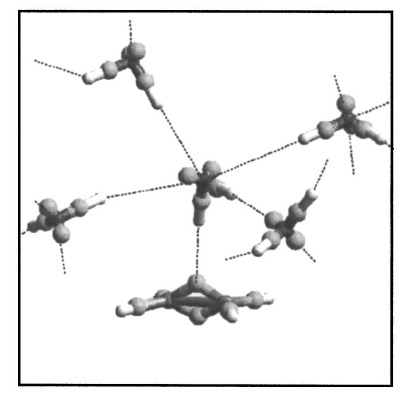
The structure and dynamics of aqueous solvation of ethanol and ethylene are studied by density functional theory based Car–Parrinello molecular dynamics. We did not find an enhancement of the structure of the hydrogen bonded network of hydrating water molecules. Both ethanol and ethylene can easily be accommodated in the hydrogen-bonded network of water molecules without altering its structure. This supports the conclusion from recent neutron diffraction experiments that there is no hydrophobic hydration around small hydrophobic groups. Analysis of the electronic charge distribution using Wannier functions shows that the dipole moment of ethanol increases from 1.8 D to 3.1 D upon solvation, while the apolar ethylene molecule attains an average dipole moment of 0.5 D. For ethylene, we identified configurations with pi-H bonded water molecules, that have rare fourfold hydrogen-bonded water coordination, yielding instantaneous dipole moments of ethylene of up to 1 D. The results provide valuable information for the improvement of empirical force fields, and point out that for an accurate description of the aqueous solvation of ethanol, and even of the apoler ethylene, polarizable force fields are required. |
|
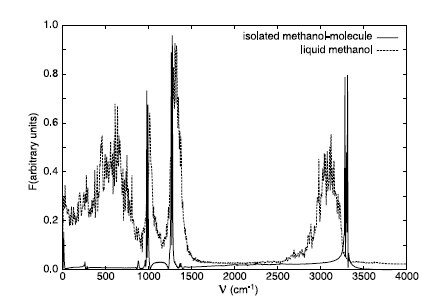
We present a density-functional theory based molecular-dynamics study of the structural, dynamical, and electronic properties of liquid methanol under ambient conditions. The calculated radial distribution functions involving the oxygen and hydroxyl hydrogen show a pronounced hydrogen bonding and compare well with recent neutron diffraction data, except for an underestimate of the oxygen-oxygen correlation. We observe that, in line with infrared spectroscopic data, the hydroxyl stretching mode is significantly red-shifted in the liquid. A substantial enhancement of the dipole moment is accompanied by significant fluctuations due to thermal motion. Our results provide valuable data for improvement of empirical potentials. |
|
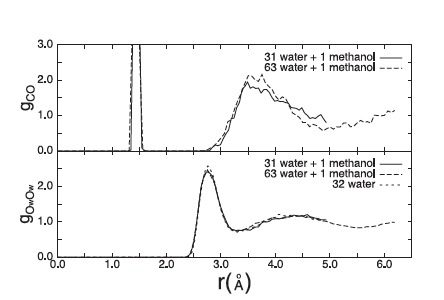
We studied the hydration of a single methanol molecule in aqueous solution by first-principle DFT-based molecular dynamics simulation. The calculations show that the local structural and short-time dynamical properties of the water molecules remain almost unchanged by the presence of the methanol, confirming the observation from recent experimental structural data for dilute solutions. We also see, in accordance with this experimental work, a distinct shell of water molecules that consists of about 15 molecules. We found no evidence for a strong tangential ordering of the water molecules in the first hydration shell. |
|
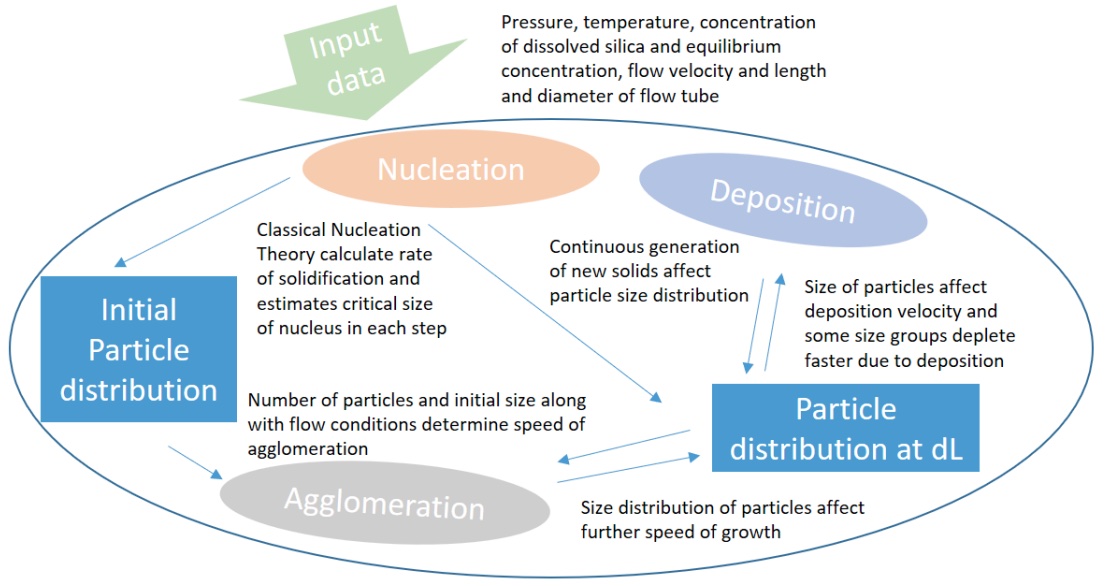
A model that can be used to quantify silica deposition from superheated depressurized steam is developed. Classical nucleation theory, agglomeration and deposition onto a wall are combined in a numerical model that calculates the concentration profile, the particle size distribution and the deposition in a flow through a pipeline after a sudden increase in supersaturation. The work presented here is an important step in understanding the mechanisms governing precipitation from supercritical and superheated steam as produced by deep geothermal wells drilled in magmatic areas. The power potential in such systems can be significantly higher than for conventional systems if utilized efficiently. The presented results can be applied to improve industrial designs and decrease energy costs. The model predicts the amount of precipitation along a pipe at various supersaturations, and the resulting deposition velocity in a straight pipe correlates fairly well with recent experimental results. There is a high number of nanocolloids formed close to the pressure reduction position, where deposition is at its maximum. Downstream, larger agglomerates develop, and deposition decreases as the number of particles and the overall concentration decreases. The local maximum deposition rate increases with increasing supersaturation. The calculations show that deposition mechanisms are as important as the chemical process of solidification when estimating where and how most material will be deposited. The rapid formation and deposition of solids predicted indicate that depressurization along with solid capture could be used to rid fluid of silica. |
|
|
The dynamics of the DNA denaturation is studied using the Peyrard-Bishop-Dauxois model. The denaturation rate of double stranded polymers decreases exponentially as a function of length below the denaturation temperature. Above Tc, the rate shows a minimum, but then increases as a function of length. We also examine the influence of sequence and solvent friction. Molecules having the same number of weak and strong base-pairs can have significantly different opening rates depending on the order of base-pairs. |
|
|
In the publication by Das T. and Chakraborty S., the authors claim to solve a problem of equilibrium statistical physics by increasing damping in a Langevin simulation, i.e. by changing the dynamics. This is a fundamental flaw which though forms the basis of the work. Increasing the friction coefficient may change how fast the phase space is explored, but, if the simulations are performed properly, it cannot alter the equilibrium distribution. The modification introduced by the authors apparently solves the problem of the divergence of <y> as the simulations start from a closed initial condition instead of using a set of initial conditions distributed over the whole phase space. The large increase of damping considerably slows down the exploration of phase space in the calculation, so much that the system stays in the vicinity of the closed initial configuration. To conclude, the simulation approach applied in this work and other related studies is not well founded in terms of statistical physics and uses the PBD model in situations where it cannot be expected to be valid. The apparent signatures of premelting in are nothing more than a memory effect of chosen initial conditions and do not hold when they are scrutinized by more accurate calculations. |
|
|
Benham and Singh commented on our letter. This was the second comment on our letter in Phys. Rev. Lett. but from a completely different viewpoint than that of the first comment by Choi et al. Benham and Singh state that superhelical stress, generated by proteins, is required to generate the transcription bubble. They also discuss their model that includes this superhelical stress. They further state that without it considerable bubble formation is negligible. We answer that they might well be right on this point, but that we did not study bubble formation in vivo. We studied thermally induced bubbles using our method to test the hypothesis of Choi et al. This hypothesis stated that thermally induced bubbles already carry a fingerprint of biological active sites. Our results did not support this hypothesis contrary to earlier less accurate MD results. However, Choi et al commented that their experimental evidence is sufficient for reasserting their hypothesis. So the main disagreement is not between us and Benham and Singh, but between Benham and Singh and Choi et al. |
|
The local opening of DNA is an intriguing phenomenon from a statistical-physics point of view, but is also essential for its biological function. For instance, the transcription and replication of our genetic code cannot take place without the unwinding of the DNA double helix. Although these biological processes are driven by proteins, there might well be a relation between these biological openings and the spontaneous bubble formation due to thermal fluctuations. Mesoscopic models, like the Peyrard-Bishop-Dauxois (PBD) model, have fairly accurately reproduced some experimental denaturation curves and the sharp phase transition in the thermodynamic limit. It is, hence, tempting to see whether these models could be used to predict the biological activity of DNA. In a previous study, we introduced a method that allows to obtain very accurate results on this subject, which showed that some previous claims in this direction, based on molecular-dynamics studies, were premature. This could either imply that the present PBD model should be improved or that biological activity can only be predicted in a more complex framework that involves interactions with proteins and super helical stresses. In this article, we give a detailed description of the statistical method introduced before. Moreover, for several DNA sequences, we give a thorough analysis of the bubble-statistics as a function of position and bubble size and the so-called l-denaturation curves that can be measured experimentally. These show that some important experimental observations are missing in the present model. We discuss how the present model could be improved. |
|
|
Choi et al commented on our letter that criticizes the simulation results of their paper Nucl. Acids Res. : DNA directs its own transcription. Choi et al. admit that our results are more accurate, but believe that their experimental results are still sufficient evidence for their hypothesis. We respond that the main aim of our article was the method that is orders of magnitude faster than MD or MC. That this method could clear up some ambiguous simulation results on a hot topic shows the importance of our method. Whether the experimental evidence is still sufficient to support the hypothesis is still an open question. However, see also the other comment to our letter by Benham and Singh who attack the hypothesis by Choi et al. in a comment to our letter. |
|
|
For certain classes of mathematical models, we developed a smart numerical method that makes expensive molecular dynamics or Monte Carlo simulations redundant. We used our technique to study the formation of bubbles in double-stranded DNA, an issue that is notoriously difficult to simulate with MD or MC. We obtained results with an almost analytical precision with only 4 hours CPU time. For comparison, an equally accurate MD simulation would last 200 years on the same computer. Thanks to this method we were able to falsify previously published MD results by another group (Choi et al: DNA directs its own transcription). The previous publications claimed evidence for a surprising hypothesis that the information on thermally induced bubbles (without the presence of proteins) can very accurately predict biological active sites. Our publication initiated a vigorous debate between supporters and opponents of the original hypothesis, which resulted in two Comments on our article. However, our method and results were never put in question. Our article was also advertised as the Scientific Highlight at the website of the CECAM institute. |
|
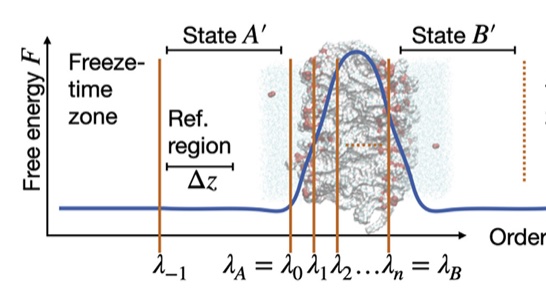
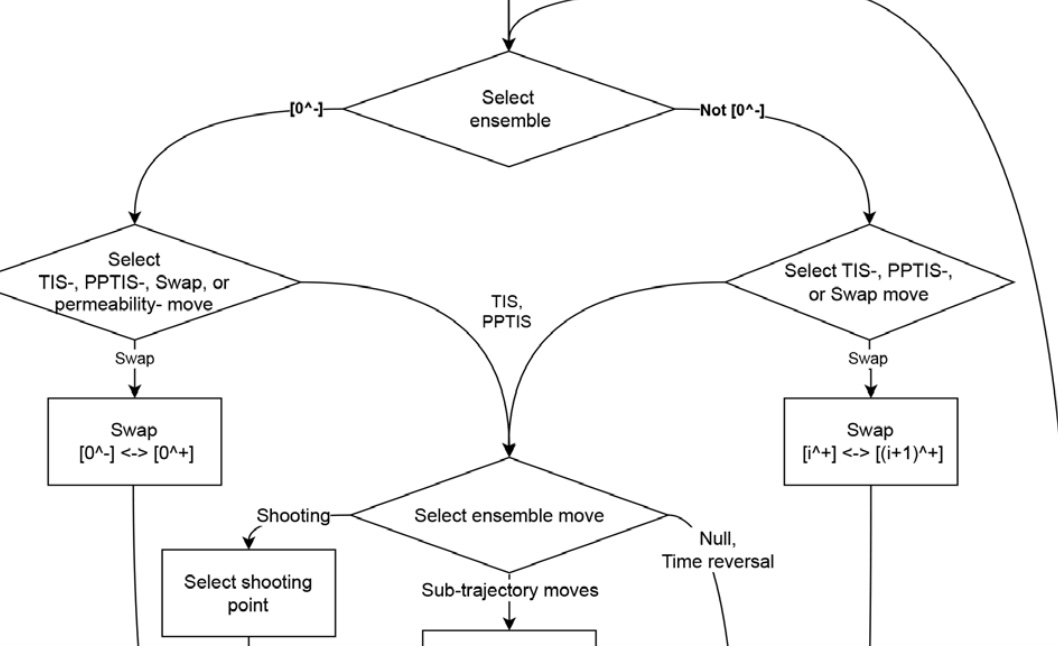
We present and discuss the advancements made in PyRETIS 3, the third instalment of our Python library for an efficient and user-friendly rare event simulation, focused to execute molecular simulations with replica exchange transition interface sampling (RETIS) and its variations. Apart from a general rewiring of the internal code towards a more modular structure, several recently developed sampling strategies have been implemented. These include recently developed Monte Carlo moves to increase path decorrelation and convergence rate, and new ensemble definitions to handle the challenges of long-lived metastable states and transitions with unbounded reactant and product states. Additionally, the post-analysis software PyVisa is now embedded in the main code, allowing fast use of machine-learning algorithms for clustering and visualising collective variables in the simulation data. |
|
The minimal-basis iterative stockholder (MBIS) and restrained electrostatic potential (RESP) methods were applied to examine the effects of edges and of nitrogen and boron dopants on the atomic partial charges of neutral and charged graphene flakes. The results provided the parameters to fit a second-order atom-condensed Kohn-Sham DFT model (ACKS2), accurately determining the partial charges, the dipole and local electric fields in large graphene flakes with negligible cost. Our approach can lead to improvements of graphene force fields in charged conditions and guide the design of media for catalytic applications. |
|
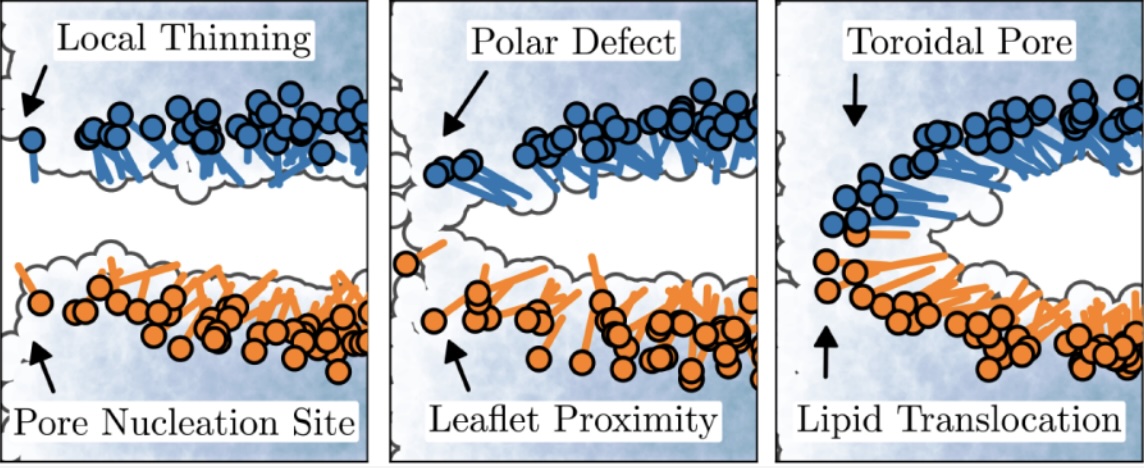
Pore formation in lipid bilayers plays a vital role in membrane fusion, transport, and signaling. Yet, its detailed mechanism remains elusive due to the limitations of conventional simulation methods. To overcome this, we apply a newly developed path sampling technique, the asynchronous and infinite swap version of Replica Exchange Transition Interface Sampling (∞RETIS), to study pore formation in a dimyristoylphosphatidylcholine (DMPC) bilayer modeled with the CHARMM36m force field. Our results reveal a sequence of tightly coupled events: pore nucleation sites are determined by earlystage thinning, and the progress into a metastable pore requires a combination of polar defects and close proximity between lipids across opposite leaflets. Using Inf-init, an initiation protocol based on ∞RETIS, rare trajectories can be generated starting directly from equilibrium simulations. Inf-init and ∞RETIS simulations reveal that lipid flip-flop occurs exclusively via local membrane thinning, and pore closure often results in asymmetric lipid distributions. |
|
Coalescence is a critical phenomenon in separation and transport processes. An improved understanding of coalescence can enhance current models predicting emulsion stability and separation. We here report a combined experimental and simulation study to investigate the thin film prior to its rupture in a coalescence event. Optical tweezers measured the influence of ions and of surface-active agents on coalescence time and the forces acting between colliding oil droplets. Molecular simulation described the composition and constituent distribution of the thin films in systems comparable with the ones investigated via optical tweezers. We identify a potential relationship between the disruption of the electrical double layer and the formation of nanocrystals with the thin film breakage times and depletion forces. |
|
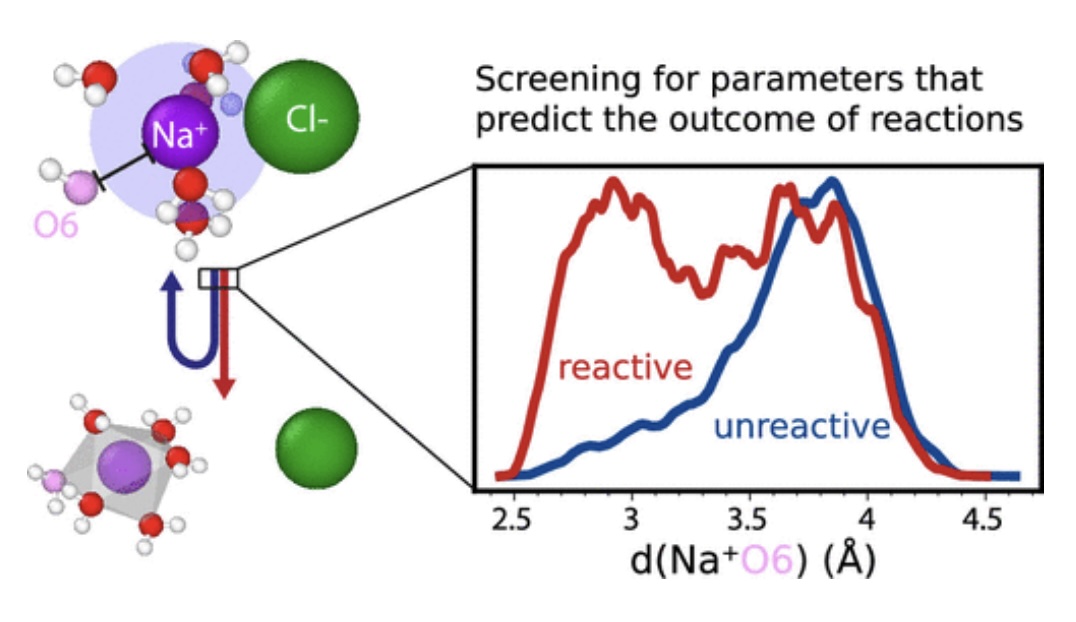
Utilizing the replica exchange transition interface sampling (RETIS) technique, we simulated the dynamics of sodium chloride dissociation in water. Subsequently, the resulting trajectories were analyzed using predictive power analysis (PPA), enabling the identification and quantification of collective variables (CVs) capable of forecasting the reaction occurrence. We improved the robustness of the PPA method by incorporating the Savitzky-Golay (SG) filter on integrated histograms, effectively avoiding the limitations associated with binning. Applying this adapted PPA method, the previously designed solvent parameters and distances from the index invariant distance matrix were assessed. This revealed that the sixth closest oxygen to sodium serves as an equally effective predictor as the best complex solvent parameter. The latter, however, required more knowledge and human intuition as an input for its design, while the former provided such intuition purely as an output. Through a comparable analysis, the chloride solvation shell appears to contain less predictive information. Employing a linear combination of several CVs can further enhance predictability, albeit at the expense of a reduced human interpretability. |
|
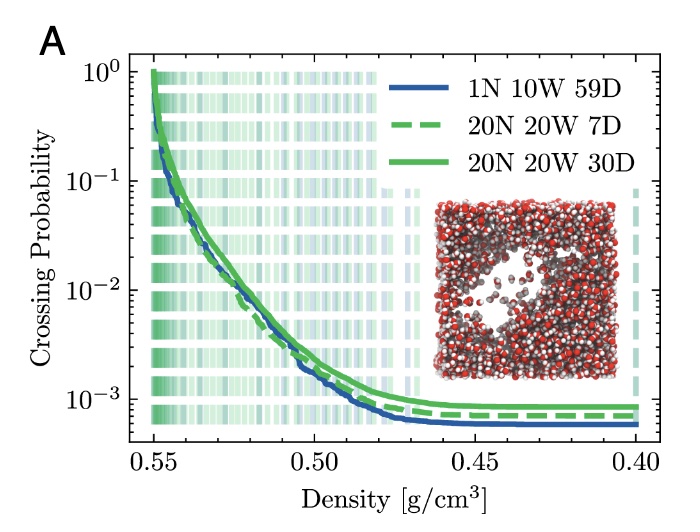
Capturing rare yet pivotal events poses a significant challenge for molecular simulations.Path sampling provides a unique approach to tackle this issue without altering thepotential energy landscape or dynamics, enabling recovery of both thermodynamicand kinetic information. However, despite its exponential acceleration compared tostandard molecular dynamics, generating numerous trajectories can still require a longtime. By harnessing our recent algorithmic innovations—particularly subtrajectorymoves with high acceptance, coupled with asynchronous replica exchange featuringinfinite swaps—we establish a highly parallelizable and rapidly converging pathsampling protocol, compatible with diverse high-performance computing architectures.We demonstrate our approach on the liquid–vapor phase transition in superheatedwater, the unfolding of the chignolin protein, and water dissociation. The latter,performed at the ab initio level, achieves comparable statistical accuracy within days,in contrast to a previous study requiring over a year. |
|
Molecular dynamics (MD) and Monte Carlo (MC) have long coexisted as two main independent branches of molecular simulation. In the late eighties, however, algorithms based on the combination of both were created such as hybrid Monte Carlo which uses large MD steps as MC moves. An entirely different kind of combination emerged a decade later via the transition path sampling (TPS) method in which MD trajectories are not just part of the MC move, but also form the state space being sampled. Algorithms like replica exchange transition interface sampling (RETIS) exploit this idea to compute reaction rates via a series of TPS simulations. RETIS yields results identical to hypothetical long MD runs, but with exponentially reduced computation time. This perspective describes the RETIS method and discusses recent and future advancements that will enable the study of even longer molecular timescales with reasonable computational resources. |
|
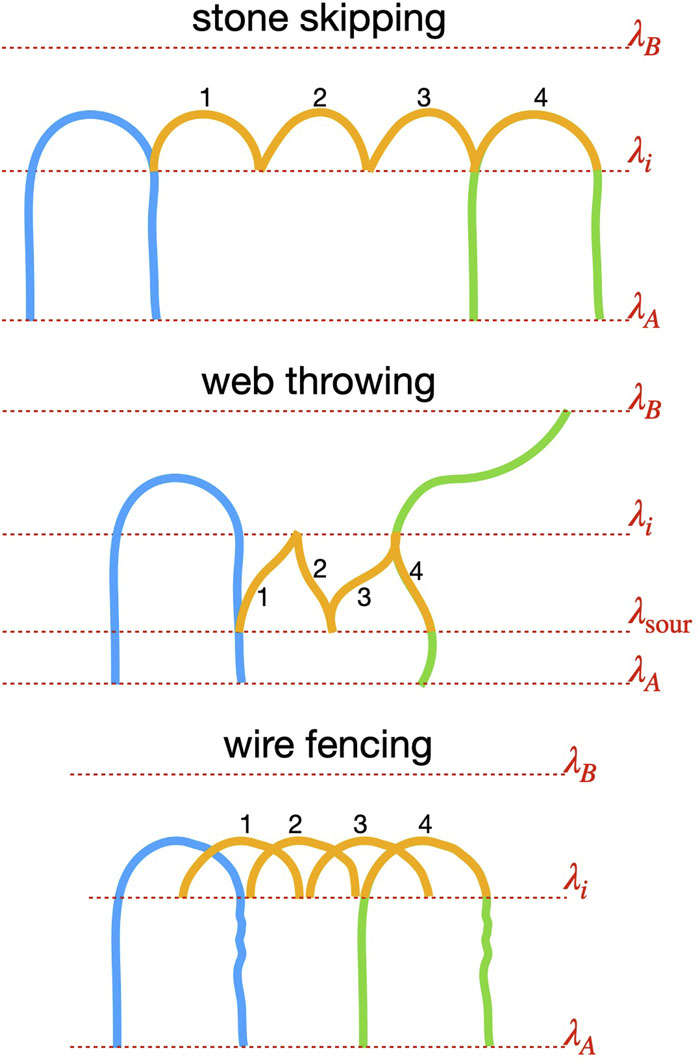
Path sampling allows the study of rare events, such as chemical reactions, nucleation, and protein folding, via a Monte Carlo (MC) exploration in path space. Instead of configuration points, this method samples short molecular dynamics (MD) trajectories with specific start- and end-conditions. As in configuration MC, its efficiency highly depends on the types of MC moves. Since the last two decades, the central MC move for path sampling has been the so-called shooting move in which a perturbed phase point of the old path is propagated backward and forward in time to generate a new path. Recently, we proposed the subtrajectory moves, stone-skipping (SS) and web-throwing, that are demonstrably more efficient. However, the one-step crossing requirement makes them somewhat more difficult to implement in combination with external MD programs or when the order parameter determination is expensive. In this article, we present strategies to address the issue. The most generic solution is a new member of subtrajectory moves, wire fencing (WF), that is less thrifty than the SS but more versatile. This makes it easier to link path sampling codes with external MD packages and provides a practical solution for cases where the calculation of the order parameter is expensive or not a simple function of geometry. We demonstrate the WF move in a double-well Langevin model, a thin film breaking transition based on classical force fields, and a smaller ruthenium redox reaction at the ab initio level in which the order parameter explicitly depends on the electron density. |
|
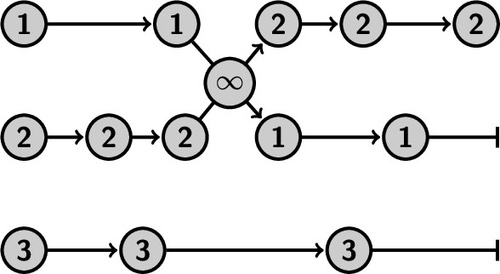
We developed a replica exchange method that is effectively parallelizable even if the computational cost of the Monte Carlo moves in the parallel replicas are considerably different, for instance, because the replicas run on different types of processor units or because of the algorithmic complexity. To prove detailed-balance, we make a paradigm shift from the common conceptual viewpoint in which the set of parallel replicas represents a high-dimensional superstate, to an ensemble-based criterion in which the other ensembles represent an environment that might or might not participate in the Monte Carlo move. In addition, based on a recent algorithm for computing permanents, we effectively increase the exchange rate to infinite without the steep factorial scaling as a function of the number of replicas. We illustrate the effectiveness of this replica exchange methodology by combining it with a quantitative path sampling method, replica exchange transition interface sampling (RETIS), in which the costs for a Monte Carlo move can vary enormously as paths in a RETIS algorithm do not have the same length and the average path lengths tend to vary considerably for the different path ensembles that run in parallel. This combination, coined ∞RETIS, was tested on three model systems. |
|
|
Using molecular dynamics and path sampling techniques we investigated the effect of pressure and defects in the wurtzite to rock salt transition in cadmium selenide (CdSe). In the pressure range 2-10 GPa, rate constants of transition are in the order of 10-23 to 105 s-1 for the transformation of a relatively small wurtzite crystal consisting of 1024 atoms with periodic boundary conditions. The transition paths predominantly evolve through an intermediate 5-coordinated structure, as reported before, though its typical lifetime within the transition paths is particularly long in the intermediate pressure range (4-6 GPa). The defects were created by removing Cd-Se pairs from an otherwise perfect crystal. The removals were either selected fully randomized or grouped in clusters (cavity creation). We find that the rate of transition due to the defects increases by several orders of magnitude even for a single pair removal. This is caused by a change in the transition mechanism that no longer proceeds via the intermediate 5-coordinated structure, when defects are present. Further, the cavity creation yields a lower rate than the fully randomized removal. |
|
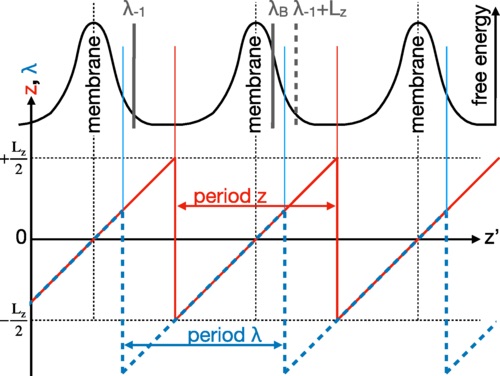
Permeation of compounds through membranes is important in biological and engineering processes, e.g., drug delivery through lipid bilayers, anesthetics, or chemical reactor design. Simulations at the atomic scale can provide insight in the diffusive pathways and they give estimates of the membrane permeability based on counting membrane transitions or on the inhomogeneous solubility-diffusivity model described by the Smoluchowski equation. For many permeants, permeation through a membrane is too slow to gather sufficient statistics with conventional molecular dynamics simulations, i.e., permeation is a rare event. Recent attempts to improve the description of the dynamics of such rare permeation events have been based on milestoning, which allows the study of processes at timescales beyond those achievable by straightforward molecular dynamics. The approach is not relying on an overdamped description, but, still, it uses a Markovian approximation which is only valid for small permeants that are not disruptive to the membrane structure. To overcome this fundamental limitation, we show here how replica exchange transition interface sampling (RETIS) can effectively be used on this problem by deriving an effective set of equations that relate the outcome of RETIS simulations and the permeability coefficient. In addition, we introduce two new path Monte Carlo (MC) moves specifically for permeation dynamics, that are used in combination with the ordinary path generating moves, which considerably increase the efficiency. The advantage of our method is that it gives exact results, identical to brute force molecular dynamics, but orders of magnitude faster. |
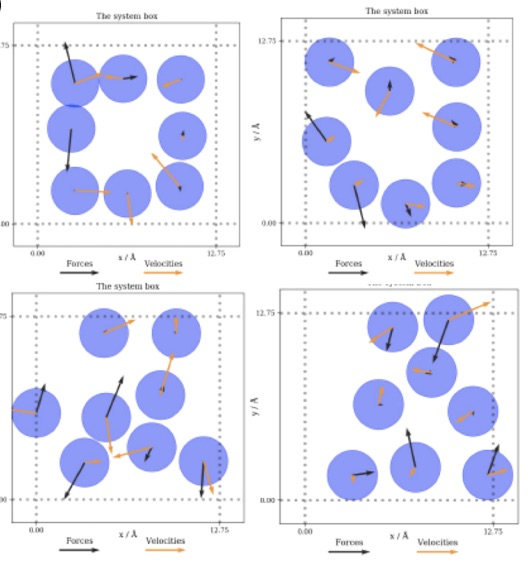
|
It is quite challenging to learn complex mathematical algorithms used in molecular simulations, stressing the importance of using the most advanta- geous teaching methods. Ideally, individuals should learn at their pace and deal with tasks fitting their levels. Web‐based exercises make it possible to tailor every small step of the learning process, but this requires continuous monitoring of the learner. Differentiation based on the scores after the first round of common tasks can be demotivating for all students, as they will experience the initial set of tasks as being either too easy or too hard. We designed two tests, a self‐monitoring test and a rapid test (RT) in which the students explained equations relating to the current topic. The first test was aimed to see if the students were able to evaluate their own level of knowledge, whereas the RT was aimed to find a fast way to determine the level of the students. We compared both tests with traditional measures of knowledge and used a relatively new method, which was originally designed for the analysis of molecular simulation data, to interpret the results. Based on this analysis, we concluded that self‐evaluation, rather than an RT, is a valuable tool to automatically steer individual students through a tree of web‐based exercises to match their skill levels and interests. |

|
The algorithmic development in the field of path sampling has made tremendous progress in recent years. Although the original transition path sampling method was mostly used as a qualitative tool to sample reaction paths, the more recent family of interface-based path sampling methods has paved the way for more quantitative rate calculation studies. Of the exact methods, the replica exchange transition interface sampling (RETIS) method is the most efficient, but rather difficult to implement. This has been the main motivation to develop the open-source Python-based computer library PyRETIS that was released in 2017. PyRETIS is designed to be easily interfaced with any molecular dynamics (MD) package using either classical or ab initio MD. In this study, we report on the principles and the software enhancements that are now included in PyRETIS 2, as well as the recent developments on the user interface, improvements of the efficiency via the implementation of new shooting moves, easier initialization procedures, analysis methods, and supported interfaced software. |
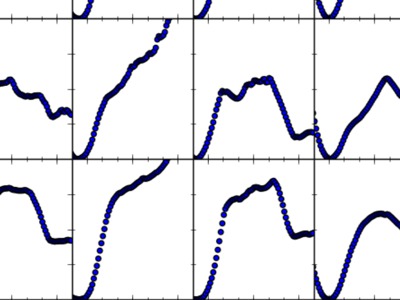
|
The exchange of methane with carbon dioxide in gas hydrates at the deep ocean has been suggested as a manner harvest methane while at the same time store carbon dioxide. Ex- perimental evidence suggest that this process is facilitated if gas mixtures are used instead of pure carbon dioxide. We studied the free energy barriers for diffusion of methane, car- bon dioxide, nitrogen, and hydrogen in the sI hydrate structure using molecular simulation techniques. Cage hops between neighboring cages were considered with and without a wa- ter vacancy and with a potential inclusion of an additional gas molecule in either the initial or final cage. Our results give little evidence for enhanced methane and carbon dioxide diffusion if nitrogen is present as well. However, the inclusion of hydrogen seem to have a substantial effect as it diffuses rapidly, can easily enter occupied cages, while reducing the barriers for the gas molecules that co-occupy a cage with hydrogen. |
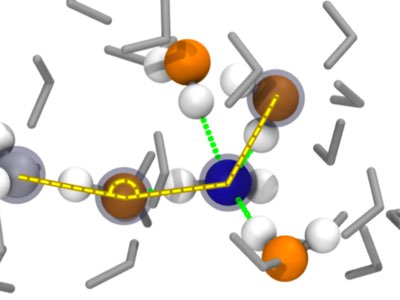
|
The pH of liquid water is determined by the infrequent process in which water molecules split into short-lived hydroxide and hydronium ions. This reaction is difficult to probe experimentally and challenging to simulate. One of the open questions is whether the local water structure around a slightly stretched OH bond is actually initiating the eventual breakage of this bond or that this event is driven by a global ordering that involves many water molecules far away from the reaction center. Here, we have investigated the self-ionization of water at room temperature by rare event ab initio molecular dynamics and obtained autoionization rates and activation energies in good agreement with experiments. Based on the analysis of thousands of molecular trajectories, we identified a couple of local order parameters and show that if a bond stretch occurs when all these parameters are around their ideal range, the chance for the first dissociation step (double proton jump) increases from 10-7 till 0.4. Understanding these initiation triggers might ultimately allow the steering of chemical reactions. |
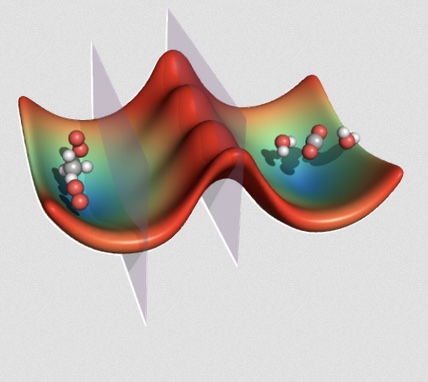
|
Transition Path Sampling techniques are becoming common approaches in the study of rare events at the molecular scale. More efficient methods, such as transition interface sampling (TIS) and replica exchange transition interface sampling (RETIS), allow the investigation of rare events, e.g. chemical reactions and structural/morphological transitions, in a reasonable computational time. Here, we present pyretis, a Python library for performing TIS and RETIS simulations. pyretis directs molecular dynamics (MD) simulations in order to sample rare events with unbiased dynamics. pyretis is designed to be easily interfaced with any molecular simulation package and in the present release, it has been interfaced with GROMACS and CP2K, for classical and ab initio MD simulations, respectively. |
|
|
Nearly 20 years ago transition path sampling (TPS) emerged as an alternative method to free energy based approaches for the study of rare events such as nucleation, protein folding, chemical reactions, and phase transitions. TPS performs effectively Monte Carlo (MC) with relatively short molecular dynamics trajectories, with the advantage of not having to alter the actual potential energy surface nor the underlying physical dynamics. Although the TPS approach also introduced a methodology to compute reaction rates, this approach was for a long time considered theoretically attractive, providing the exact same results as extensively long molecular dynamics simulations, but still expensive for most relevant applications. With the increase of computer power and improvements in the algorithmic methodology, quantitative path sampling is finding applications in more and more areas of research. In particular, the transition interface sampling (TIS) and the replica exchange TIS (RETIS) algorithms have, in turns, improved the efficiency of quantitative path sampling significantly, while maintaining the exact nature of the approach. Also, open-source software packages are making these methods, for which implementation is not straightforward, now available for a wider group of users. In addition, a blooming development takes place regarding both applications and algorithmic refinements. Therefore, it is timely to explore the wide panorama of the new developments in this field. This is the aim of this article, which focuses on the most efficient exact path sampling approach, RETIS, as well as its recent applications, extensions and variations. |
|
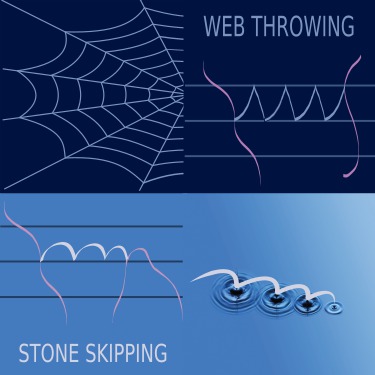
Many relevant processes in chemistry, physics, and biology are rare events from a computational perspective as they take place beyond the accessible time scale of molecular dynamics (MD). Examples are chemical reactions, nucleation, and conformational changes of biomolecules. Path sampling is an approach to break this time scale limit via a Monte Carlo (MC) sampling of MD trajectories. Still, many trajectories are needed for accurately predicting rate constants. To improve the speed of convergence, we propose two new MC moves, stone skipping and web throwing. In these moves, trajectories are constructed via a sequence of subpaths obeying super-detailed balance. By a reweighting procedure, almost all paths can be accepted. Whereas the generation of a single trajectory becomes more expensive, the reduced correlation results in a significant speedup. For a study on DNA denaturation the increase was found to be a factor 12. |
|
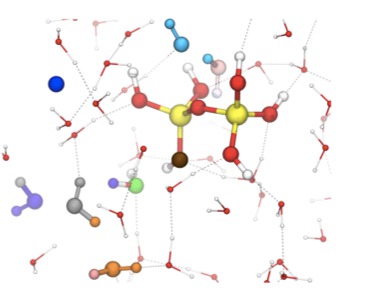
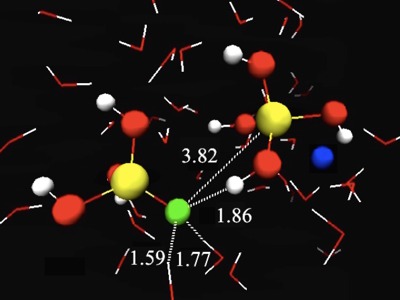
A replica exchange transition interface sampling (RETIS) study combined with Born-Oppenheimer molecular dynamics (BOMD) is used to investigate the dynamics, thermodynamics and mechanism of the early stages of the silicate condensation process. In this process, two silicate monomers, of which one anion species, form a negatively charged five-coordinated silicate dimer. In a second stage, this dimer can fall apart again, forming the original monomers, or release a water molecule into the solution. We studied the association and dissociation reaction in the gas phase, and the dissociation and water removal step in the aqueous phase. The results on the aqueous phase dissociation suggest two possible mechanisms. The breakage of the bond between the intermediate oxygen and the five-coordinated silicon is sometimes accompanied with a proton transfer. After the dissociation into silicate monomers, the anionic monomer is either the previously four-coordinated silicon or the previously five-coordinated silicon depending on whether the hydrogen transfer occurs or not. Our results show that the mechanism with proton transfer is highly predominant. The water removal simulations also show two possible mechanisms distinguished by the proton transfer reaction path. The proton transfer can either occur via a direct or via a water mediated reaction step. The calculations reveal that although both mechanisms contribute to the water removal process, the direct proton transfer is slightly favorable and occurs roughly in six out of ten occasions. This is the first time ever that the RETIS approach is applied in combination with Ab Initio molecular dynamics. In this study, we revealed some crucial mechanistic steps in the process of silica oligomerization which would not be possible to detect by any other method as they tend to disrupt the spontaneous dynamics of the system. |
|
We studied silica dimerization reactions in the gas and aqueous phase by density functional theory (DFT) and reactive force fields based on two parameterizations of ReaxFF. For each method (both ReaxFF force fields and DFT) we performed constrained geometry optimizations, which were subsequently evaluated in single point energy calculations using the other two methods. Standard fitting procedures will typically compare the force field energies and geometries with those from quantum mechanical data after a geometry optimization. The initial configurations for the force field optimization are usually the minimum energy structures of the ab initio database. Hence, the ab initio method dictates which structures are being examined and force field parameters are being adjusted in order to minimize the differences with the ab initio data. As a result, this approach will not exclude the possibility that the force field predicts stable geometries or low transition states which are realistically very high in energy and, therefore, never considered by the ab initio method. Our analysis reveals the existence of such unphysical geometries even at unreactive conditions where the distance between the reactants is large. To test the effect of these discrepancies, we launched molecular dynamics simulations using DFT and ReaxFF and observed spurious reactions for both ReaxFF force fields. Our results suggest that the standard procedures for parameter fitting need to be improved by a mutual comparative method. |
|
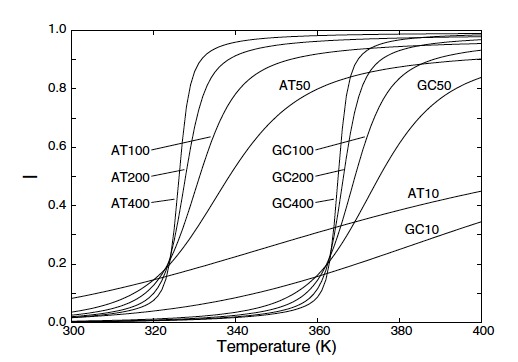
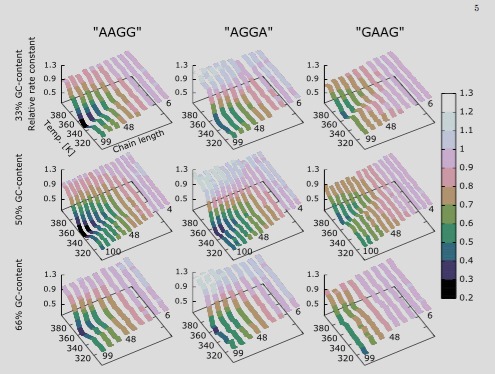
Using rare event simulation techniques, we calculated DNA denaturation rate constants for a range of sequences and temperatures for the Peyrard-Bishop-Dauxois (PBD) model with two different parameter sets. We studied a larger variety of sequences compared to previous studies that only consider DNA homopolymers and DNA sequences containing an equal amount of weak AT- and strong GC-base pairs. Our results show that, contrary to previous findings, an even distribution of the strong GC-base pairs does not always result in the fastest possible denaturation. In addition, we applied an adaptation of the PBD model to study hairpin denaturation for which experimental data are available. This is the first quantitative study in which dynamical results from the mesoscopic PBD model have been compared with experiments. Our results show that present parameterized models, although giving good results regarding thermodynamic properties, overestimate denaturation rates by orders of magnitude. We believe that our dynamical approach is, therefore, an important tool for verifying DNA models and for developing next generation models that have higher predictive power than present ones. |
|
We develop a new method combining replica exchange transition interface sampling with two distinct potential energy surfaces. The method can be used to combine different levels of theory in a simulation of a molecular process (e.g. a chemical reaction) and it can serve as a dynamical version of QM-MM, connecting classical dynamics with Ab Initio dynamics in the time domain. This new method, which we coin QuanTIS, could be applied to use accurate but expensive density functional theory based molecular dynamics for the breaking and making of chemical bonds, while the diffusion of reactants in the solvent are treated with classical force fields. We exemplify the method by applying it to two simple model systems (an ion dissociation reaction and a classical hydrogen model) and we discuss a possible extension of the method in which classical force field parameters for chemical reactions can be optimized on the fly. This article is freely available by open-access. |
|
We studied the adsorption of chiral mixtures of lactic acid in several zeolites. All zeolite systems showed either no selectivity or heteroselectivity in which the minority enantiomer is adsorbed by a higher fraction than its fraction in the reservoir. Analysis of the mechanism showed that none of the previously identified origins of enantioselective adsorption of scalemic mixtures applies to lactic acid. However, based on the lack of any ordered distribution in the adsorbed phase we postulate a new mechanism that is likely to be very generic for chiral adsorption processes that proceed via chaotic packing of the adsorbate molecules. The new mechanism can explain several characteristics of the adsorption data and hints on new prospective separation methods with a high potential for farmaceutical applications. Our surprising finding shows that chaos is sometimes more effective than order! |
|
We used molecular simulation techniques to analyze the enantioselective adsorption of chiral polar molecules in achiral zeolites. As a representative chiral molecule, CHClFBr was used in this study. We analyzed the adsorption of racemic and scalemic mixtures of these molecules into different zeolites topologies such as MFI and MEL, both structures with intersecting channels, and FER and TON, frames with nonintersecting channels. In contrast to previous findings on apolar molecules, we found that cations are not essential for imposing heteroselective adsorption, and homoselective adsorption was never observed. We derived the mechanism behind the selectivity, which turns out to be fundamentally different from that observed for apolar adsorbates. Our model is able to explain why apparently similar zeolite topologies can still have strong differences in their adsorption behavior. |
|
I discuss the reactive flux (RF) method and the more recent algorithms that originate from the transition path sampling (TPS) methodology. These comprise the transition interface sampling (TIS) and the replica exchange TIS (RETIS), which are successive improvements on the way reaction rates were determined in the original TPS algorithm. Partial path TIS (PPTIS) is an approximative approach in order to reduce the simulated path length for the case of diffusive barrier crossings. PPTIS is similar to Milestoning, that was developed simultaneously and independently from PPTIS. For nonequilibrium systems, the forward flux sampling (FFS) was designed. This method is based on the TIS formalism, but does not require prior knowledge on the phasepoint density. All these methods have in common that they aim to simulate true molecular dynamics trajectories at a much faster rate than naive brute force molecular dynamics. I discuss the advantages and disadvantages of different path sampling methodologies. In the end, I compare all the methods by applying them on a simple, though tricky, test system. The outcome illustrates some important pitfalls for the nonequilibrium methods (all splitting-based methods like FFS, WEB dynamics, Russian-Roulette, ...) that have no easy solution and show that caution is necessary when interpreting their results. |
|
We propose in this paper a generic model of a nonstandard aggregation mechanism for self-assembly processes of a class of materials involving the mediation of intermediates consisting of a polydisperse population of nanosized particles. The model accounts for a long induction period in the process. The proposed mechanism also gives insight on future experiments aiming at a more comprehensive picture of the role of self-organization in self-assembly processes. |
|
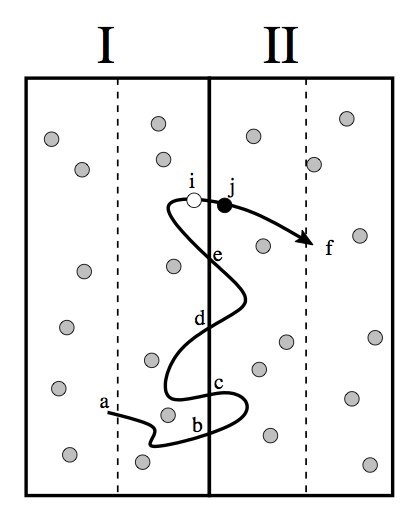
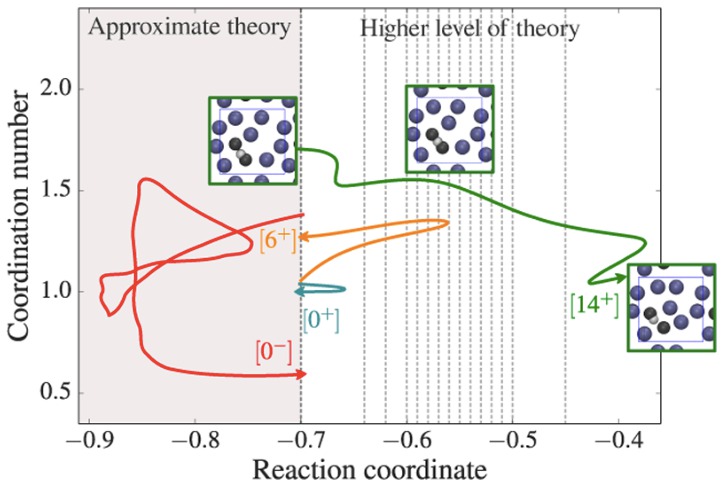
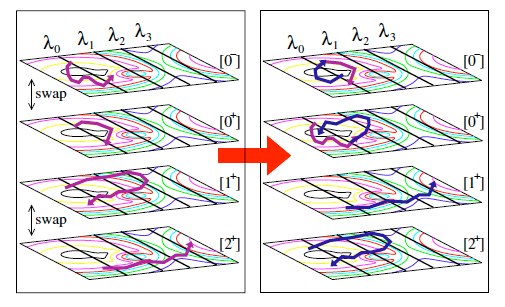
Due to the time scale problem, rare events are not accessible by straight forward molecular dynamics. The presence of multiple reaction channels complicates the problem even further. The feasibility of the standard free energy based methods relies strongly on the success in finding a proper reaction coordinate. This can be very difficult task in high-dimensional complex systems and even more if several distinct reaction channels exist. Even if a proper reaction coordinate can be found, ergodic sampling will be a challenge. In this article, we discuss the recent advancements of path sampling methods to tackle this problem. We argue why the path sampling methods, via the transition interface sampling technique, is less sensitive to the choice of reaction coordinate. Moreover, we review a new algorithm, parallel path swapping, that can dramatically improve the ergodic sampling of trajectories for the multiple reaction channel systems. |
|
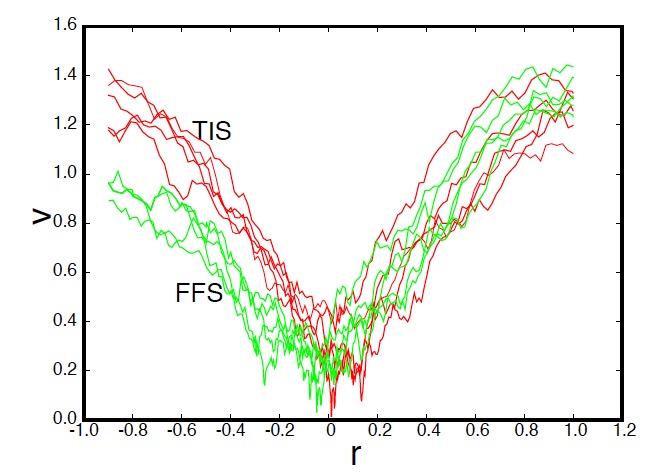
The efficiency of path sampling simulations can be improved considerably using the approach of path swapping. For this purpose, we devise a new algorithmic procedure based on the transition interface sampling technique. In the same spirit of parallel tempering, paths between different ensembles are swapped, but the role of temperature is here played by the interface position. We test the method on the denaturation transition of DNA using the Peyrard-Bishop-Dauxois model. We find that the new algorithm gives a reduction of the computational cost by a factor of 20. |
|
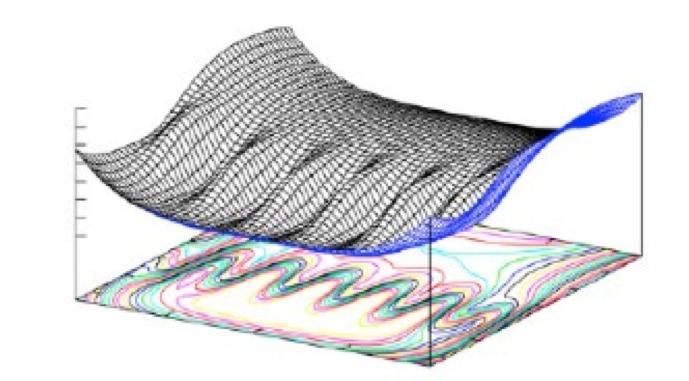
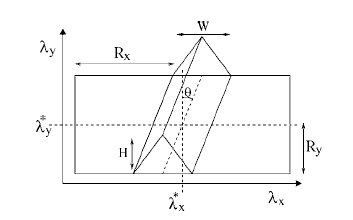
We analyze the efficiency of different methods for the calculation of reaction rates in the case of a simple two-dimensional analytical benchmark system. Two classes of methods are considered: the first is based on the free energy calculation along a reaction coordinate and the calculation of the transmission coefficient, the second on the sampling of dynamical pathways. We give scaling rules for how this efficiency depends on barrier height and width, and we hand out simple optimization rules for the method-specific parameters. We show that the path sampling methods, using the transition interface sampling technique, become exceedingly more efficient than the others when the reaction coordinate is not the optimal one. |
|
In a previous paper of one of us [Europhys. Lett. 59, 330-336 (2002)] the validity of Greene's method for determining the critical constant of the standard map (SM) was questioned on the basis of some numerical findings. Here we come back to that analysis and we provide an interpretation of the numerical results, by showing that the conclusions of that paper were wrong as they relied on a plausible but untrue assumption. Hence no contradiction exists with respect to Greene's method. We show that the previous results, based on the expansion in Lindstedt series, do correspond to the critical constant but for a different map: the semi-standard map (SSM). For such a map no Greene's method analog is at disposal, so that methods based on Lindstedt series are essentially the only possible ones. Moreover, we study the expansion for two simplified models obtained from the SM and SSM by suppressing the small divisors. We call them the simplified SM and simplified SSM, respectively; the first case turns out to be related to Kepler''s equation after a proper transformation of variables. In both cases we give an analytical solution for the radius of convergence, that represents the singularity in the complex plane closest to the origin. Also here, the radius of convergence of the simplified SM turns out to be lower than that of the simplified SSM. However, despite the absence of small divisors these two radii are lower than those of the true maps (i.e., of the maps with small divisors) when the winding number equals the golden mean. Finally, we study the analyticity domain and, in particular, the critical constant for the two maps without small divisors. The analyticity domain turns out to be a perfect circle for the simplified SSM (as for the SSM itself), while it is stretched along the real axis for the simplified SM, yielding a critical constant which is larger than its radius of convergence. |
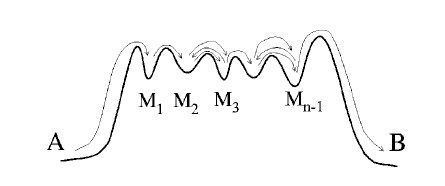
|
We introduce a path sampling method for the computation of rate constants for complex systems with a highly diffusive character. Based on the recently developed transition interface sampling (TIS) algorithm this procedure increases the efficiency by sampling only parts of complete transition trajectories. The algorithm assumes the loss of memory for diffusive progression along the reaction coordinate. We compare the new partial path technique to the TIS method for a simple diatomic system and show that the computational effort of the new method scales linearly, instead of quadratically, with the width of the diffusive barrier. The validity of the memory loss assumption is also discussed. |